Où, quand, comment le chien a été domestiqué ?
Où, quand, comment le chien a été domestiqué ?
On sait que le chien est la première espèce animale à avoir été domestiquée par l’Homme. Bien avant le Néolithique, on trouve des restes canins, associés à des occupations humaines, dont les caractéristiques morphologiques permettent d’appeler « chien » et non plus loup. Les plus anciennes de ces traces remontent à 14 000 ans, alors que l’Homme moderne était encore un chasseur-cueilleur. Que le chien ait été une aide certaine pour la chasse ne fait aucun doute ; mais cela soulève une autre question : 14 000 ans ? Et pourquoi pas avant ?
Et là, les débateurs s’écharpent. Malheureusement, aucun argument convaincant ne ressort du débat. Le loup a pu être domestiqué et garder pendant longtemps une morphologie voisine de ses congénères sauvages qui rend indistinguable chiens et loups du Paléolithique sur des données purement morphométriques. Et d’ailleurs, des loups bizarres ont été trouvés dans des grottes européennes, qui ont conduits certains archéozoologues à dater cette domestication au Paléolithique supérieur (Germonpré et al. 2009; Ovodov et al. 2011; Germonpré et al. 2012). Trouvaille critiquée par d’autres archéozoologues qui concluent que la présence d’une plus grande variabilité phénotypique des loups du Paléolithique était déjà connue et n’implique pas que certains avatars soient des chiens (Boudadi-Maligne and Escarguel 2014).
Et la génétique, que dit-elle ? Beaucoup de choses, avec à peu près autant de contradictions (Freedman et al. 2014). On constate déjà que la majeure partie des races de chiens actuels dérivent d’une sélection qui s’est produite il y a quelques centaines d’années et qui a conduit à l’existence de variétés aussi dissemblables. Si on observe le génome des chiens domestiques actuels pour rechercher des événements démographiques, on trouve trace principalement de cette sélection très forte – tandis que les traces de la domestication (ou des domestications) sont trop ténues pour aboutir à des résultats concluants.
On peut comparer les génomes de chiens à ceux de loups pour calculer la date du dernier ancêtre commun. On aboutit ainsi à une date très ancienne, mais qui ne correspond pas du tout à un événement de domestication, mais à celle de séparation de deux lignées de loups : celle qui conduit aux loups actuels et celle à partir de laquelle le chien a été domestiqué. Domestication qui a pu se produire des dizaines de milliers d’années après.
D’autant plus que la paléogénétique montre que une forte perte de diversité génétique (mitochondriale) chez les loups entre le Paléolithique Supérieur et la période actuelle (Thalmann et al. 2013) : il est fort possible que la lignée de loup à partir de laquelle le chien a été domestiquée ait disparu.
Et l’affaire est compliquée par le fait que chiens et loups appartiennent à la même espèce : ils sont encore interféconds et les flux génétiques entre les deux groupes, domestiqués et sauvages, n’ont jamais vraiment cessé.
Et enfin, un seul génome de loup ancien a pour l’instant été publié (Skoglund et al. 2015): il s’agit d’un loup qui n’est ni ancestral aux loups actuels, ni ancestral aux chiens actuels.
Pour tenter de reconstituer l’histoire de la domestication du chien, une publication récente dans PNAS (Shannon et al. 2015) se tourne vers les « chiens de village », des chiens commensaux des humains, vivant à proximité des habitations, ni vraiment sauvages, ni vraiment domestiqués, et dans la reproduction desquels l’Homme n’intervient pas. Avec un peu de chance, ces chiens auraient gardé une trace de l’histoire démographique plus ancienne ?
Pour faire cette étude, sur plus de 500 chiens de village, et 5000 chiens de « race », les auteurs ne se sont pas tournés vers du séquençage de génome complet, mais ont utilisé des puces qui permettent d’enquêter sur 200 000 variants génétiques présents chez le chien.

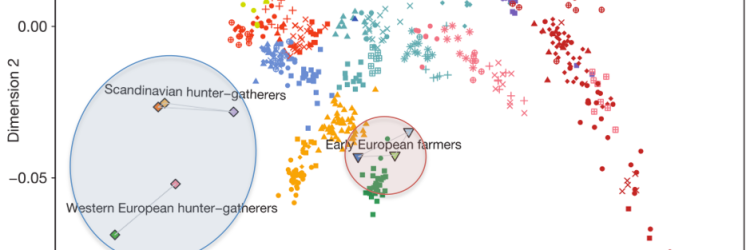
Analyse en composante principale des variants génétiques de chiens actuels (Adapté de Shannon et al. 2015)
Qu’observe-t-on ? Déjà que les données sont complexes et que la représentation choisie par les auteurs n’est pas des plus faciles (représenter en cercle pleins et noirs les chiens de village européens les rend difficile à distinguer des races de chiens en cercles vides et noirs …). La diversité génétique des chiens de village est difficile à corréler à la géographie, puisque la première composante de l’ACP, qui explique un quart de la variabilité des données) ne permet pas d’établir un lien avec la géographie ; il faut attendre la deuxième composante pour trouver une phylogéographie, de l’Afrique vers l’Asie et l’Europe.
La seconde information est plus simple à voir : certaines races de chiens ressemblent beaucoup, génétiquement, aux chiens de village des régions dont elles sont issues. Une surprise ? Pas franchement.
Troisième information : visiblement, les chiens de villages de régions assez éloignées (les Amériques, la Polynésie) sont proches des chiens de villages européens, sans doute grâce aux migrations humaines.
Cependant, cette approche montre que l’idée de s’intéresser aux chiens de village était pertinente, et qu’ils sont porteurs d’une plus grande diversité génétique que les chiens de race.
Les auteurs se sont ensuite intéressés au déséquilibre de liaison. Le déséquilibre de liaison intervient quand deux allèles sont plus fréquemment regroupés qu’attendu aléatoirement en fonction de la distance physique et génétique qui les regroupe. Plus des allèles sont éloignés sur le génome, plus il est rare qu’ils restent associés. Or, cette valeur de déséquilibre de liaison (ou LD) est soumise à de nombreux effets démographiques (et à la sélection : deux allèles qui « marchent bien » ensemble auront tendance à rester soudés) : en particulier, des changements de taille de population et des migrations vont avoir un effet sur le LD. Ainsi, le passage par un goulot d’étranglement va augmenter le LD, tandis que des flux génétiques sous forme de migrations vont le diminuer.
Passage par un goulot d’étranglement ? N’est ce pas exactement ce à quoi on s’attend en cas de domestication ?

Valeurs des déséquilibres de liaisons selon les régions d’origine des chiens de village. Les valeurs présentées en A correspondent au LD à 0,005 cM. (Adapté de Shannon et al. 2015)
Et effectivement, les auteurs observent un LD inférieur chez les loups que chez les chiens … montrant que les seconds ont subit un goulot d’étranglement drastique. Parmis les chiens de villages, les auteurs ont ensuite essayé de trouver dans quelle région du monde cet événement s’est-il produit ? En partant du principe que les populations de chiens qui proviennent d’une autre région que la région initiale sont eux même un sous-échantillon, et ont subi une diminution encore plus forte de la taille de leur population, ils ont regardé quels étaient les chiens avec le LD le plus faible … et ont trouvé que les chiens d’Asie Centrale correspondaient le mieux à cette définition.
CQFD ? On a trouvé la région où le chien a été domestiqué, quelque part entre la Sibérie et le Népal ?
Je n’en suis pas certaine … Déjà, parce que la comparaison des LD manque cruellement de statistiques : 0,71, est-ce significativement différent de 0,72 ou de 0,73 ? Je ne suis pas convaincue… De plus, pourquoi choisir le LD à 0,005 cM, et non celui à 0,02 cM (dans ce cas, c’est au Vietnam que la domestication s’est produite) ou celui à 0,2 cM (qui pointe vers l’Inde) ? Je pense que cette incertitude des résultats est la preuve que l’histoire génétique du chien est très complexe, très difficile à modéliser simplement, et que leur papier prouve simplement que les chiens actuels descendent d’un goulot d’étranglement qui s’est produit quelque part vers l’Asie.
Et pourquoi dater la domestication du chien à la domestication des chiens actuels ? Si des domestications se sont produites plus précocement, même si elles n’ont pas donné de descendants dans le présent, ne comptent-elles pour rien ? L’idée que l’Homme pouvaient agir sur la nature, sur la reproduction d’animaux sauvages, et sélectionner des traits qui l’avantagent lui, et non les animaux, ne nait-elle pas de la première domestication ?
Au final, je conclurai comme les auteurs : il est définitivement très difficile de dire quoique ce soit à partir des chiens actuels, et une plongée dans la diversité génétique des chiens, et des loups, et de ceux dont on ne connaît pas très bien le statut, du passé est nécessaire. Mais j’y joindrai une remarque supplémentaire : difficile de dire à partir d’un génome si c’est un toutou ou un loup à qui on a affaire. Des études génomiques pour déterminer les régions du génome impliquées dans la domestication sont elles aussi très précieuses.
Bibliographie
Boudadi-Maligne M, Escarguel G. 2014. A biometric re-evaluation of recent claims for Early Upper Palaeolithic wolf domestication in Eurasia. J. Archaeol. Sci. 45:80–89.
Freedman AH, Gronau I, Schweizer RM, et al. 2014. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 10:e1004016.
Germonpré M, Lázničková-Galetová M, Sablin M V. 2012. Palaeolithic dog skulls at the Gravettian Předmostí site, the Czech Republic. J. Archaeol. Sci. 39:184–202.
Germonpré M, Sablin M V., Stevens RE, Hedges REM, Hofreiter M, Stiller M, Després VR. 2009. Fossil dogs and wolves from Palaeolithic sites in Belgium, the Ukraine and Russia: osteometry, ancient DNA and stable isotopes. J. Archaeol. Sci. 36:473–490. A
Ovodov ND, Crockford SJ, Kuzmin Y V, Higham TFG, Hodgins GWL, van der Plicht J. 2011. A 33,000-year-old incipient dog from the Altai Mountains of Siberia: evidence of the earliest domestication disrupted by the Last Glacial Maximum. PLoS One 6:e22821.
Shannon LM, Boyko RH, Castelhano M, et al. 2015. Genetic structure in village dogs reveals a Central Asian domestication origin. Proc. Natl. Acad. Sci.:6–11.
Skoglund P, Ersmark E, Palkopoulou E, Dalén L. 2015. Ancient Wolf Genome Reveals an Early Divergence of Domestic Dog Ancestors and Admixture into High-Latitude Breeds. Curr. Biol. :1–5.
Thalmann O, Shapiro B, Cui P, et al. 2013. Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs. Science (80-. ). 342:871–874.